|
||
|
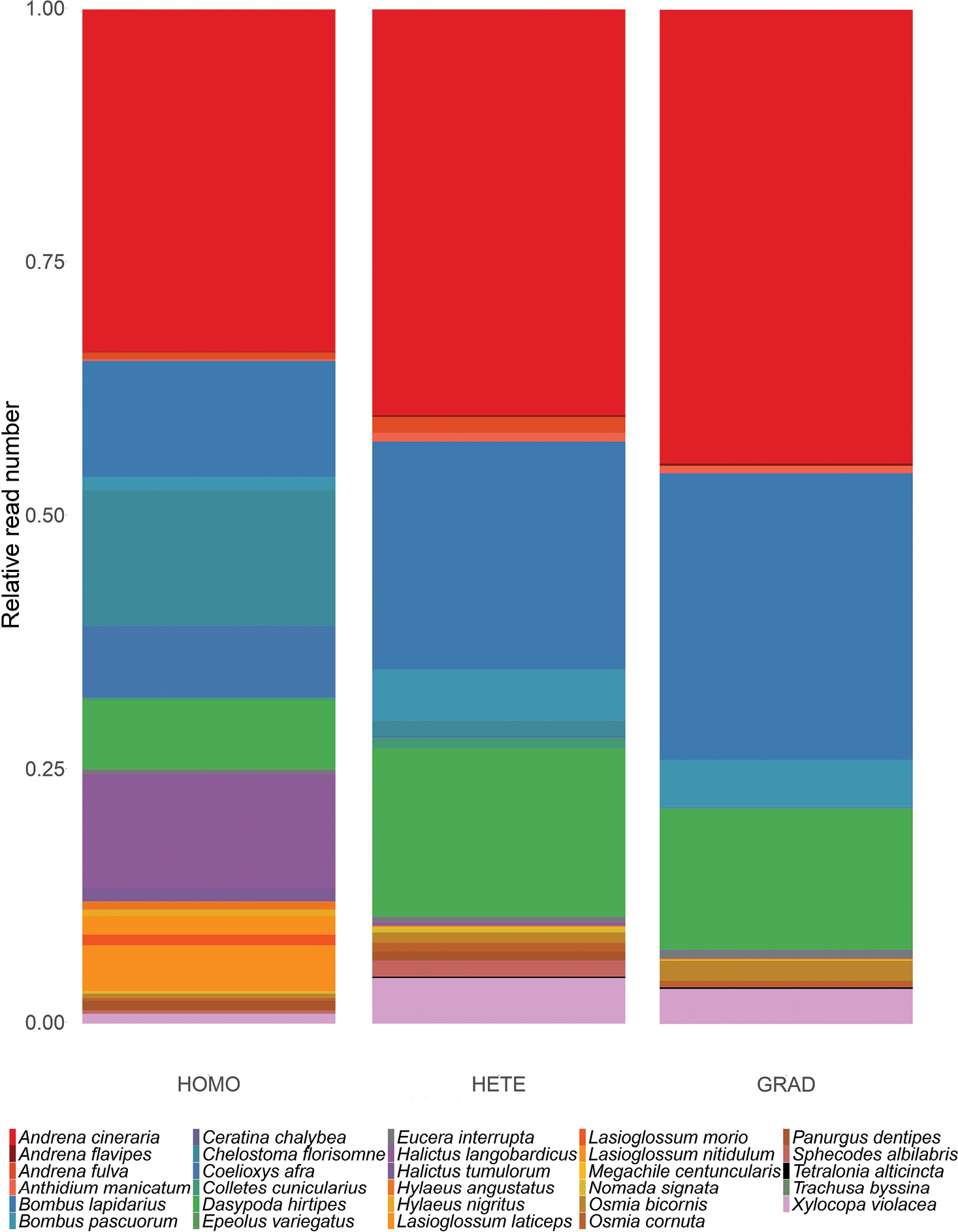
Proportion of sequencing reads per species in each mock community. Each community was assembled from the DNA of the same 29 specimens (25 fresh ones and 4 dry ones), all of them belonging to different species; the HOMO community was based on equimolar pools of the individual DNA extractions; the HETE community was assembled by pooling 1 ul of isolated DNA from a single leg from each specimen and the GRAD community was made by modifying the original concentrations of each species based on their concentration categories in order to exaggerate existing biomass differences. Relative read numbers were obtained by averaging absolute read numbers from all three replicates of each mock community and then correcting by the total number of reads in each treatment. A significant difference was found only between GRAD and HOMO (see text for details). |